
Introduction to a Common Pathway in Cancer Biology
Wading through Biological Complexity... For Fun


Through my past few years of undergrad studying Biochemistry, I’ve memorized my fair share of cellular pathways such as the one above. It’s brutal. There’s so many moving parts, x dimerizing y to activate z which then binds to a… What’s crazy to think about is that in the world of drug discovery, not only do we need to identify the right targets to focus on, but we have to find the right compounds with the right features (ADME-Tox and more) that are capable of acting upon those targets to hopefully cure us of said disease. It’s an extremely complicated process.
This led me down an interesting path of trying to better understand how AI and machine learning can be used for more effective drug discovery. To highlight the complexity of cellular pathways and our crazy attempts at trying to understand disease mechanisms (which sometimes resorts to sheer luck) within this vast realm of biological complexity, I have a dug up an old paper (with slight modifications) to share with you today. This was from a paper I wrote for one of my cancer pharmacology courses back in January 2020 when I was studying abroad at the National University of Singapore.
It provides a brief overview of the Akt/PI3K signalling pathway as well as takes you through some of the historic attempts we’ve had at targeting specific parts within this pathway. You’ll see that it a constant back and forth. We think we found the perfect drug for the perfect target in this pathway, only to find out that the drug has extremely harmful side effects or the target is not the right one. It’s a bit different from the posts I’ve released in the past but hope you enjoy nonetheless!
Introduction to the Akt/PI3K Signalling Pathway
The activation of this pathway begins at the cell membrane when growth signals/factors (i.e. Insulin) bind to a receptor tyrosine kinase (RTK) (Garcia-Echeverria and Sellers, 2008). This causes dimerization of the RTK’s and enables cross-phosphorylation of tyrosine residues in the intracellular domains (Zhang, et al. 2020). Once this occurs, PI3K is recruited and PI3K phosphorylates PIP2 (bounded to cell membrane) into PIP3, thus seeing an increase in PIP3 levels. The role of PIP3 is to recruit PDK1, an important molecule in this pathway as it leads to the phosphorylation and activation of Akt. Akt has 2 sites available for phosphorylation, one at Thr308 and another at Ser473, these sites only become exposed when Akt undergoes a conformational change after binding to PI3K. Then, PDK1 can directly phosphorylate at Thr308, or it can activate mTOR complex 2 which can phosphorylate Akt at Ser473 (“AKT Signalling Pathway…”, 2017). There are several downstream pathways that can now be activated by Akt. One pathway to highlight would be the activation of mTOR. Regardless of the specific downstream pathway, the activation of the Akt/Phosphoinositide 3-kinase (PI3K) signalling pathway controls cell cycle progression, cell survival, protein synthesis, cell growth and angiogenesis, thus it is clear to see why aberrant activation and mutations within this pathway can commonly lead to various types of cancer (Garcia-Echeverria and Sellers, 2008). For the purpose of this paper, several molecules out of this highly complex pathway will be chosen and analyzed, those molecules will be: PI3K, PDK1, Akt and mTOR.
PI3K Structure and Possible Mutations
PI3K is a family of enzymes that are categorized into 3 classes, Class I, II and III (Garcia-Echeverria and Sellers, 2008). Mutations in Class I have been proven to be associated with cancer due to the role it plays in cell growth, proliferation and survival while Class II and III, which plays a role in intracellular membrane trafficking, has not been proved to be linked to any diseases yet (Jean et al., 2014). Class I phosphorylates PIP2 to PIP3 in vivo and is composed of subclass 1A and 1B.
Subclass 1A has p85 as a regulatory subunit and p110 as a catalytic subunit which exists in 3 forms, p110α, p110β and p110δ (also known as PI3K α, β, δ), subclass 1B is mainly composed of p110γ (Jean et al., 2014). Mutations typically occur in the p110 catalytic subunit at p110α and p110β which are encoded by oncogene PIK3CA and PIK3CB, respectively (Whale et al., 2017). Bowel cancer, HER2-positive breast cancer and ovarian cancer typically arise from mutation to PIK3CA, whereas minimal links have been found for cancer associated with mutations in PIK3CB. The p85 regulatory subunit contains various isoforms as well, recently, p85α and p55γ have been proved to be related to cancer due to mutations in the respective oncogenes Pik3r1 and Pik3rd (Whale et al., 2017).
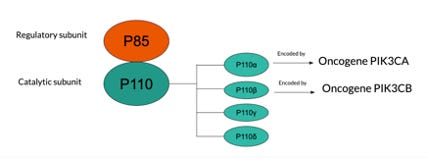
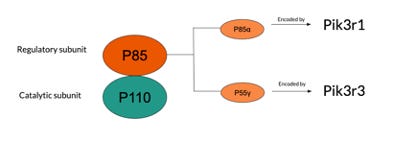
(Figure 1. Visual description of the structural components of PI3K and the various oncogenes susceptible to mutation)
PI3K as a Target for Inhibition
Since PI3K plays a vital role early on in the activation of the PI3K/Akt pathway, the development of PI3K inhibitors has been widely studied to prevent aberrant activation of this pathway. Wortmannin and LY294002 are two pan-PI3K inhibitors that played a key role as a research tool to better understand the role of PI3K’s and how they cause cancer (Garcia-Echeverria and Sellers, 2008). Wortmannin was an irreversible inhibitor while LY294003 was a reversible, ATP-competitive PI3K modulator that also had anti-angiogenic properties, it inhibited the expression of VEGF (Yamori and Kong, 2008). Although both sounded promising in theory, they displayed quite unfavorable pharmaceutical properties (liver toxicity, low bioavailability, poor solubility) and never made it to clinical trials (Garcia-Echeverria and Sellers, 2008).
PX-886
PX-886 is a semi-synthetic viridin derivative essentially modified from Wortmannin. This drug showed to be promising; it was much more stable, it inhibited PI3K longer than Wortmannin, displayed anti-tumour activity and had reduced liver toxicity as a side effect (Garcia-Echeverria and Sellers, 2008). However, it did display some newfound side effects such as a decrease in body weight, leukocytosis and most notably, hyperglycemia (high glucose levels in the blood) with a decrease in glucose tolerance in the body.
This side effect involving glucose, occurs due to the role in which the Akt/PI3 pathway also plays in glucose uptake in the body. Activated Akt will phosphorylate AS160 (a negative regulator of GLUT4 translocation) and inhibit it. Therefore, since Akt has inhibited AS160, the cell is now able to deposit GLUT4 in the membrane which eventually allows glucose to pass through the cell membrane from the bloodstream into cell where it can then undergo glycolysis (Świderska et al., 2018). At the same time, activated Akt also phosphorylates glycogen synthesis kinase 3 (GSK3) and inactivates/inhibits it. GSK3 phosphorylates glycogen synthase to inactive it. In this case, activated Akt is inhibiting an inhibitor (GSK3), subsequently leading to the activation of glycogen synthase, a key enzyme recorded for glycogen synthesis (excess glucose in the body is stored as glycogen) (Świderska et al., 2018). Therefore, the inhibition of PI3K will lead to the inactivation of Akt and can cause an array of side effects surrounding glucose intolerance and hyperglycemia.
SF-1126
SF-1126 is a promising drug, it is the water-soluble prodrug of LY294002 (Mahadevan, et al., 2013). It is known to inhibit all forms of PI3K as well as mTOR and DNA-PK. Other advantages it has over LY294002 is better pharmacokinetic properties, high distribution across tumour tissues and increased solubility. Furthermore, SF1126 has been shown to display anti-tumour and anti-angiogenic activity through numerous xenograft models (Singh et al., 2014).
NVP-BEZ235
Another promising drug is NVP-BEZ235 which is currently undergoing phase I and II trials. NVP-BEZ235 is a derivative of imidazoquinolinone that acts both as a pan inhibitor of PI3K and mTOR (Hsu et al., 2018). In cellular settings, this drug has displayed tremendous antiproliferative activity through the blocking of the PI3K signalling complex, inducing arrest in the G1 stage of the cell cycle (Garcia-Echeverria and Sellers, 2008). Furthermore, it has impact on a variety of various tumour cell lines and this activity has also been replicated within in-vivo models of human cancer (Garcia-Echeverria and Sellers, 2008). NVP-BEZ325 has also been shown to play a role in reducing tumour interstitial fluid pressure and vascular permeability, these are promising characteristics displayed by the drug that prove its effectiveness in treating cancer (Schnell et al., 2008).
A high tumor interstitial fluid pressure creates the mechanical factors that enable certain growth parameters of the tumor to grow in size. In addition, the mechanical stretch enabled by a high tumor interstitial fluid pressure has been proven to trigger cell proliferation (Hofmann et al., 2006). As a result, this negatively impacts the delivery and uptake of cancer drugs leading to diminished therapeutic effects when attempting to treat the tumor. The growth and metastasis of cancer occurs through angiogenesis, it is during this phase that cancer cells increase vascular permeability. Increasing vascular permeability allows the wall of blood vessels to increase the flow of small molecules (nutrients, ions, water, etc.) entering a specific area, providing the necessary nutrients for a tumor to grow (Roman et al., 2013). At the same time, an increase in vascular permeability creates blood flow disturbances which can minimize the efficacy and accuracy of any conventional drugs that are being delivered intravenously (Simon et al., 2008). Cancer biomarkers are used to detect cancer, it signals the growth and progression of the cancer, reflects the efficacy of any drugs that have been used to treat the cancer and is a great diagnostic, prognostic or detection tool (Goossens et al. 2015). Vascular permeability can be measured noninvasively and be used as a cancer biomarker to indicate the long-term efficacy of the treatment using NVP-BEZ235 (Simon et al., 2008). Although NVP-BEZ235 has been tested in-vivo and has excellent results when used in treating cancer (anti-growth properties and an increase in apoptosis), severe side effects were also exhibited. Some of those side effects were diarrhoea, hair loss and increase levels of both blood glucose and a liver enzyme, alanine transaminase (Netland et al., 2016). Higher levels of alanine transaminase found in the liver is known to be associated with liver damage/toxicity (Hall et al., 2012).
PDK1 Structure and Possible Mutations
PDK1 plays a crucial role in the activation of Akt in the pathway, leading to subsequent activation of downstream pathways. Theoretically, a mutation in the genes coding PDK1 can cause over expression of PDK1 resulting in the over-activation of the pathway to lead to cancer. However, research to date has not been able to identify any sort of mutations to the PDK1 gene, instead, over expression of PDK1 is more commonly attributed to mutations upstream of it (i.e. PI3K and PTEN mutations) (Maurer et al., 2009). Recent research papers have highlighted PDK1 as a promising therapeutic target, it has been proven that alteration of PDK1 is a critical component in PI3K signalling in breast cancer while also playing a role in regulating cell migration (Gagliardi et al., 2018).
Targeting PDK1
UCN-01
UCN-01 is currently in clinical trials and has shown to inactivate and dephosphorylate Akt thereby inhibiting the activation of the rest of the downstream pathway (Sato et al., 2002). UCN-01 does not act directly on Akt, rather it is a potent and non-selective inhibitor of PDK1 (Sato et al., 2002). The future use of UCN-01 is questionable due to its toxic side effects. A recent report published in 2012 outlining the details of the phase I and II clinical trials for UCN-01 to treat patients with metastatic melanoma concluded that the drug does not have sufficient clinical activity and yielded quite unfavorable side effects (Li et al., 2012). Some of these side effects, most likely caused by the poor selectivity of UCN-01, included: hyperglycemia, diarrhea, pulmonary toxicity, induced insulin resistance and more (Li et al., 2012).
Numerous studies that have been conducted have shown the correlation between inflammation and cancer (Rayburn et al,. 2009). Some shocking studies have revealed some insightful figures: patients with prostatitis (swelling of the prostate) are 14% more at risk for prostate cancer (Rothman et al,. 2004), patients with ulcerative colitis (chronic inflammation and ulcers in the digestive tract) are 25% more likely to get colorectal cancer (Loftus, 2006) and patients with pancreatitis (inflammation of the pancreas) and 10-20 folds more likely to get pancreatic cancer (Farrow et al., 2004). Chronic inflammation can induce the production of cytokines, a molecule responsible for stimulating blood vessel growth that can enable angiogenesis and metastasis for tumors (Hofseth et al,. 2006). Furthermore, many factors such as tissue destruction (Strukov et al,. 1984), disruption of DNA repair pathways and apoptosis inhibition (Sarkar et al,. 2006) are all characteristics of chronic inflammation that eventually enable mutations to occur creates a nutritious environment for tumours to grow.
Anti-Inflammatory Drugs
Throughout history there has been numerous anti-inflammatory drugs that have a method of action of inhibiting the family of enzymes called cyclooxygenase’s (COX) which is responsible for the production of prostaglandins (Vane et al,. 1998). Celecoxib is a drug that was mainly used for anti-inflammatory purposes as it is a COX-2 inhibitor (Zhu et al., 2004). However, Celecoxib has been shown to slightly improve anti-proliferative activity through preventing the activation of Akt (Arico et al,. 2002). The method of action is inhibiting the kinase activity of PDK1 by acting as an ATP competitor, competing for binding. Currently, Celecoxib is being further investigated in clinical trials with additional derivatives of Celecoxib such as OSU-03013, also being tested for effectiveness (Zhu et al., 2004).
BX-320 is another compound that acts as a PDK1 inhibitor by competing with ATP (Feldman et al,. 2005). Although BX-320 displays good selectivity over PKA (Sayle et al,. 2003), it has not yet entered clinical trials (Islam et al,. 2007).
Background Information on Akt
Akt is a serine threonine kinase, it might be one of the most widely research components of the Akt/PI3K pathway due to its centralized role. The activation of Akt can signal 100+ substrates downstream that all lead to factors such as, cell cycle progression, cell survival, angiogenesis (via VEGF) that cause cancer (Vara et al,. 2004). 3 isoforms of Akt have been identified, Akt1; found to be a major factor in a variety of cancers, Akt2; found to be one of the factors that cause cancer but not as common as Akt1 and Akt3, found to be primarily expressed in the brain but a solid connection between the role it plays in cancer has not yet been established (Manning et al,. 2007). All 3 of these isoforms are encoded by 3 different genes (Bellacosa et al,. 1991). Instead of mutations, we typically see a higher rate of gene amplification of Akt (commonly it’s with Akt2), that has been linked with thyroid, breast, ovarian cancers and more (Altomare et al,. 2005). The rare mutations that do occur typically happen in the PH domain of Akt1 as a point mutation, replacing glutamic acid with lysine (Kumar et al,. 2013) or G171R (Akt3) and E49K (Akt1) substitutions, also occurring in the kinase and PH domain, respectively (Askham et al,. 2010).
It is interesting to note that it is much more common to find PI3K mutations in cancer patients rather than Akt gene mutations, these are typically found at a much lower rate for cancers (Mundi et al,. 2016). In the sections analyzed above, it’s clear to see that there are numerous factors upstream of Akt that can lead to its aberrant activation. In addition to upstream factors, post-translational modifications of Akt such as: lysine modifications, acetylation and tyrosine phosphorylation have a crucial role in the hyperactivation of Akt in many cancers (Chan et al,. 2014). Finally, recent studies have shown the involvement of Akt in cancers developing acquired resistance not only against traditional chemotherapy, but also targeted therapies such as tamoxifen and trastuzumab (Sale et al,. 2007).
Akt Inhibitors
Historically, perfecting the best Akt inhibitor has been a serious challenge, there are currently no drugs that have been able to produce a single positive randomized phase III trials (Dienstmann et al,. 2014). However, there are numerous Akt inhibitors currently in clinical trials and they can generally be categorized into 4 main methods of action (Pal et al,. 2010). The first category are drugs such as BAY-1125976 and CCT128930 which target specific isoforms of Akt, in this case, Akt ½ and Akt 2, respectively (Lindsley et al,. 2007). Drugs such as AZD5363, GD-0068 and afuresetib are pan-Akt kinase inhibitors and also fall under this category. MK-2206 is the best-known drug headlining the second category: allosteric inhibitors of the Akt kinase domain. Lipid-based inhibitors are the third category, their method of action is inhibiting the generation of PIP3 from PIP2 via PI3K, thereby, indirectly inhibiting all isoforms of Akt from activating (Nemunaitis et al,. 2011). One of the most well-known drugs and the furthest along in clinical trials in this category is, perifosine (Crul et al,. 2002). Recall from the previous sections that activation of Akt must occur at the cell membrane, the final category prevents this localization (Tolcher et al,. 2009). The method of action is drugs such as PTX-200, PX-316 and triciribine which interact with the PH domain of Akt (Meuillet et al,. 2004).
MK-2206
MK-2206 is an orally administered drug and has had quite a bit of traction when it comes to clinical studies. It was discovered that MK-2206 exhibits synergy for treating nonsmall cell lung cancer with erlotinib and in breast cancer cell lines with lapatinib during preclinical studies (Hirai et al,. 2010). Although there were some positive results when treating women ranging from Stage I to III breast cancer (some fully recovering), the studies have revealed several unwanted side effects such as: rashes, gastrointestinal disturbances, myelosuppression and test abnormalities with liver function (Speranza et al,. 2015)
Perifosine
Perifosine is also an orally administered drug but has a method of action as a lipid-based inhibitor of Akt. Clinical studies have revealed tremendous synergistic activity with capecitabine that is effective in colorectal cancer cell lines (Richardson et al,. 2015) and with bortezomid and dexamethasone in the treatment of multiple myeloma (Richardson et al,. 2015). Some side effects exhibited in 26% and 18% of patients during clinical trials of 32 patients were grade III neutropenia (low levels of neutrophils, a type of white blood cell) and thrombocytopenia, respectively. More common side effects that saw over half of the patients experience was fatigue and diarrhoea (Jakubowiak et al,. 2012).
Main Components of mTOR and Possible Mutations
mTOR is also a serine threonine kinase which forms two distinct signalling complexes, mTORC1 and mTORC2 (Chao et al,. 2019). Within the two signalling complexes, there are also unique and common subunits. Common subunits are mLST8 and DEPTOR while the unique subunits to mTORC1 are PRAS40 and RAPTOR, and mSIN1, PROTOR1 and PROTOR2 for mTORC2 (Chao et al,. 2019). The main function of mTORC1 is nutrient and growth factor signalling for anabolic metabolism, it has been widely shown in research the association between hyperactive mTORC1 as a common driver in various kidney cancers (Chao et al,. 2019). The main functions of mTORC2 are to coordinate with PDK1 in order to complete the fully activation of Akt (via phosphorylation) (Chao et al,. 2019). mTOR is quite prone to mutations and these are the mutations commonly seen during aberrant activation of the Akt/PI3K pathway leading to cancer (Chao et al,. 2019). Research has shown there are approximately 570 missense mutations that are linked to 20 different cancer types with these mutations occurring at various frequencies and at different amino acid positions as well (Chao et al,. 2019).
mTOR Inhibitors
mTOR inhibitors can also be categorized into two main groups, first generation and second-generation drugs. First generation drugs (Rapamycin, Temsirolimus and Everolimus to name a few) have already been approved and are in use, however, a certain level of adverse side effects still exist. The second-generation drugs (AZD8055, INK128 and OSI027 to name a few) are in clinical trials hoping to maintain or improve effectiveness while reducing these side effects (Xie et al,. 2016).
Rapamycin (Sirolimus)
Rapamycin has been in use ever since 2000, but it was not always used in the fight against cancer. Originally, it was used as an immunosuppressant (blocking the activation of T-cells) to prevent rejection during kidney grafts (Xie et al,. 2016). Along with the protein FKBP12, rapamycin will bind to mTORC1 and not mTORC2 and essentially reduce the substrate’s access to the catalytic site of mTORC1 (Xie et al,. 2016). The method of action here is targeting the mTORC1 dimer, the rapamycin-FKBP12 complex will bind to the dimer and reduce the access and availability of the active site from 20A to 10A, in the dimer (Xie et al,. 2016). As it doesn’t affect mTORC2 in any way, the results only show the inhibition of some mTOR function but not all. An array of side effects are associated with the administration of rapamycin including: thrombocytopenia, fever, anemia, nausea, hypertriglyceridemia and more (Xie et al,. 2016), the pharmacological properties are also subpar due to its low oral bioavailability and large pharmacokinetic variability (Xie et al,. 2016). However, rapamycin has become a gateway to the development of new drugs that are analogs of rapamycin (rapalogs) that have superior characteristics and less side effects (Xie et al,. 2016).
Temsirolimus
Temsirolimus is an example of a rapalog, initially approved in 2007 to be used as treatment for renal cell carcinoma (RCC) (Xie et al,. 2016). RCC is what 90-95% of adults are diagnosed with when they have kidney cancer and the survival rates are generally quite high (Xie et al,. 2016). According to a recent report, the 5 year survival rate for stage I patients is 94% and stage II is at 79% (Xie et al,. 2016). Temsirolimus is not specific to inhibiting mTORC1 or mTORC2, rather it inhibits the activity of mTOR entirely (Xie et al,. 2016).. The method of action is similar to Rapamycin whereby Temsirolimus will form a drug-protein complex by binding to FKBP-12 that will inhibit mTOR activity and trigger cell cycle arrest at G1 phase (Xie et al,. 2016). Additionally, Temsirolimus decreases hypoxia-inducible factor (HIF) 1α, one of the regulators responsible for sustaining the survival and growth of cancer cells, which causes a reduction in the synthesis of VEGF, a crucial factor enabling angiogenesis (Xie et al,. 2016). The use of this drug has also expanded outside of just the treatment of renal cell carcinoma and is now being used to treat lymphomas as well (Xie et al,. 2016). Temsirolimus is not free from side effects as well, some of the more notable effects include a decrease lymphocytes and hemoglobin’s, and lung toxicity (Xie et al,. 2016).
INK128
Most of the clinical studies being conducted using INK128 have been using the drug to treat high-risk neuroblastoma, a type of cancer that causes nearly 15% of deaths in children who have been diagnosed with some sort of cancer (Zhang et al,. 2015). Currently, the 5 year survival rate sits at 40% for high-risk neuroblastoma, even with treatment using intense therapeutics (high-dose chemotherapy) due to drug resistance and a decrease in sensitivity (Zhang et al,. 2015). The MYCN gene has been linked to cause 20% of neuroblastoma cases, this gene plays a key role in cell growth and survival, most of the tests conducted on the efficacy of INK128 were done on MYCN-amplified cell lines (IMR32, NB-10 and NGP) (Zhang et al,. 2015). INK128 inhibits both mTORC1 and mTORC2 by acting as a small molecule, ATP-competitive inhibitor and is currently being tested in phase I clinical trials (Zhang et al,. 2015). It was discovered that in both in vitro and in vivo (xenograft mouse model) settings, INK128 can induce cell cycle arrest and apoptosis (Zhang et al,. 2015). Moreover, when used with other treatments such as doxorubicin, INK128 was able to increase the effect it had on neuroblastoma by increasing sensitivity (Zhang et al,. 2015).
Another interesting discovery during trials was the ability of INK128 to inhibit lactate production and glucose uptake in neuroblastoma cells (Zhang et al,. 2015). This is important because historic studies have outlined the association between a high uptake of glucose and production of lactate to be a key feature in cancer cells (Zhang et al,. 2015).
OSI-027
OSI-027 is another drug that has a method of action of being a small molecule, ATP-competitive inhibitor, it displays tremendous selectivity of mTORC1 and mTORC2 without affecting other components in the pathway such as PI3Kα, PI3Kβ, PI3Kγ (Shripad et al,. 2010). OSI-027 was shown to be better and inhibiting cell proliferation and inducing cell death than Rapamycin (Shripad et al,. 2010). Most of the tests being conducted were testing the efficacy of OSI-027 for the treatment of colorectal tumors (Shripad et al,. 2010).
Other Pathway Components
Aside from the PI3K, PDK, Akt and mTOR as key targets of the Akt/PI3K pathway, there are other important components that are also being investigated into how drugs can be created to inhibit their action. These 2 components are PTEN and PDK2, briefly summarized below.
PTEN
PTEN is a component of the Akt/PI3K pathway that is responsibility for promoting DNA repair and chromosome stability, furthermore, it has been identified that genomic instability arises from a loss of normal PTEN function (Dillon et al,. 2014). The most common mutation with the PTEN gene is found on chromosome 10q23, it’s associated with most of the tumors found in human cancer (Dillon et al,. 2014). PTEN exists in both they cytoplasm and nuclear form, each with a different function. They cytoplasmic form is more responsible for the regulation of PI3K/PIP3 signalling whereas nuclear PTEN has tumor suppressive function and regulates chromosome stability, apoptosis and DNA repair (Dillon et al,. 2014).
PDK2
In the pathway, PDK2 plays a role as one of the regulators of oxidative phosphorylation and glycolysis, it also plays a role in tumor maintenance and cell proliferation. Initially, the need to identify PDK2 as a component arose from the drug resistance displayed by non-small cell lung cancer to a drug called Paclitaxel (Taxol) (Sun et al,. 2017). The Warburg effect was the observation that cancer cells typically undergo aerobic glycolysis rather than oxidative phosphorylation as an energy source, even when abundant oxygen was present, was discovered in the 1920’s (Sun et al,. 2017). This is important because the relationship between Paclitaxel drug resistance and cancer is not fully understood yet and targeting the regulation of aerobic glycolysis and oxidative phosphorylation (via PDK2) could be a key to decreasing drug resistance (Sun et al,. 2017).
If you’ve made it all the way to the end, thanks for sticking it through! If you learned something from this piece, please subscribe and share with 3 friends, I would greatly appreciate it!
Please also shoot me a follow on Twitter!
References
Altomare, D. A. and Testa, J. R. (2005). Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464.
Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P et al. (2002). Celecoxib induces apoptosis by inhibiting 3-phosphoinosi- tide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem 277: 27613–27621.
Askham, J. M., Platt, F., Chambers, P. A., Snowden, H., Taylor, C. F. and Knowles, M. A. (2009). AKT1 mutations in bladder cancer: identification of a novel oncogenic mutation that can co-operate with E17K. Oncogene 29, 150–155.
Bellacosa, A., Testa, Staal, S. and Tsichlis, P. (1991). A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 254, 274–277.
Bendell, J. C., Nemunaitis, J., Vukelja, S. J., Hagenstad, C., Campos, L. T., Hermann, R. C., Sportelli, P., Gardner, L. and Richards, D. A. (2011). Randomized Placebo-Controlled Phase II Trial of Perifosine Plus Capecitabine As Second- or Third-Line Therapy in Patients With Metastatic Colorectal Cancer. Journal of Clinical Oncology 29, 4394–4400.
Bhagwat, S. V., Crew, A. P., Gokhale, P. C., Yao, Y., Kahler, J., Epstein, D. M., Wild, R. and Pachter, J. A. (2010). Abstract 4487: OSI-027, a potent and selective small molecule mTORC1/mTORC2 kinase inhibitor is mechanistically distinct from rapamycin. Experimental and Molecular Therapeutics.
Cancer Res August 15 2008 (68) (16) 6598-6607; DOI: 10.1158/0008-5472.CAN-08-1044
Chan, C. H., Jo, U., Kohrman, A., Rezaeian, A. H., Chou, P. C., Logothetis, C., & Lin, H. K. (2014). Posttranslational regulation of Akt in human cancer. Cell & bioscience, 4(1), 59. https://doi.org/10.1186/2045-3701-4-59
Chao, L. H., & Avruch, J. (2019). Cryo-EM insight into the structure of MTOR complex 1 and its interactions with Rheb and substrates. F1000Research, 8, F1000 Faculty Rev-14. https://doi.org/10.12688/f1000research.16109.1
Christian R. Schnell, Frédéric Stauffer, Peter R. Allegrini, Terence O'Reilly, Paul M.J. McSheehy, Celine Dartois, Michael Stumm, Robert Cozens, Amanda Littlewood-Evans, Carlos García-Echeverría and Sauveur-Michel Maira
Crul, M., Rosing, H., Klerk, G. D., Dubbelman, R., Traiser, M., Reichert, S., Knebel, N., Schellens,
Dienstmann, R., Rodon, J., Serra, V. and Tabernero, J. (2014). Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Molecular Cancer Therapeutics 13, 1021–1031.
Dillon, L. and Miller, T. (2014). Therapeutic Targeting of Cancers with Loss of PTEN Function. Current Drug Targets 15, 65–79.
Do, K., Speranza, G., Bishop, R., Khin, S., Rubinstein, L., Kinders, R. J., Datiles, M., Eugeni, M., Lam, M. H., Doyle, L. A., et al. (2015). Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investigational New Drugs 33, 720–728.
Ewa Świderska, Justyna Strycharz, Adam Wróblewski, Janusz Szemraj, Józef Drzewoski and Agnieszka Śliwińska (November 5th 2018). Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake, Blood Glucose Levels, Leszek Szablewski, IntechOpen, DOI: 10.5772/intechopen.80402. Available from: https://www.intechopen.com/books/blood-glucose-levels/role-of-pi3k-akt-pathway-in-insulin-mediated-glucose-uptake
Farrow, B., Sugiyama, Y., Chen, A., Uffort, E., Nealon, W., & Mark Evers, B. (2004). Inflammatory mechanisms contributing to pancreatic cancer development. Annals of surgery, 239(6), 763–771. https://doi.org/10.1097/01.sla.0000128681.76786.07
Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D et al. (2005). Novel small molecule inhibitors of 3-phosphoinositide- dependent kinase-1. J Biol Chem 280: 19867–19874
Gagliardi, P. A., Puliafito, A. and Primo, L. (2017). PDK1: At the crossroad of cancer signaling pathways. Seminars in Cancer Biology.
Garcia-Echeverria, C. and Sellers, W. R. (2008). Drug discovery approaches targeting the PI3K/Akt pathway in cancer. 1–16.
Goossens, N., Nakagawa, S., Sun, X., & Hoshida, Y. (2015). Cancer biomarker discovery and validation. Translational cancer research, 4(3), 256–269. https://doi.org/10.3978/j.issn.2218-676X.2015.06.04
Hall, P., & Cash, J. (2012). What is the real function of the liver 'function' tests?. The Ulster medical journal, 81(1), 30–36.
Hirai, H., Sootome, H., Nakatsuru, Y., Miyama, K., Taguchi, S., Tsujioka, K., Ueno, Y., Hatch, H., Majumder, P. K., Pan, B.-S., et al. (2010). MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Molecular Cancer Therapeutics 9, 1956–1967.
Hofmann, M., Guschel, M., Bernd, A., Bereiter-Hahn, J., Kaufmann, R., Tandi, C., Wiig, H., & Kippenberger, S. (2006). Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia (New York, N.Y.), 8(2), 89–95. https://doi.org/10.1593/neo.05469
Hofseth, L. J. and Ying, L. (2006). Identifying and defusing weapons of mass inflammation in carcinogenesis. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1765, 74–84.
Hsu, C., Lin, P., Tsai, Y. et al. NVP-BEZ235, a dual PI3K-mTOR inhibitor, suppresses the growth of FaDu hypopharyngeal squamous cell carcinoma and has a synergistic effect with Cisplatin. Cell Death Discovery 4, 57 (2018). https://doi.org/10.1038/s41420-018-0060-7
Islam I, Brown G, Bryant J, Hrvatin P, Kochanny MJ, Phillips GB et al. (2007a). Indoline-based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 2: optimization of BX-517. Bioorg Med Chem Lett 17: 3819–3825
J., Beijnen, J. and Huinink, W. T. B. (2002). Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. European Journal of Cancer 38, 1615–1621.
Jakubowiak AJ, Richardson PG, Zimmerman T, Alsina M, Kaufman JL, Kandarpa M, et al. Perifosine plus lenalidomide and dexamethasone in relapsed and relapsed/refractory multiple myeloma: a Phase I Multiple Myeloma Research Consortium study. Br J Haematol 2012; 158: 472–80.
Jean, S. and Kiger, A. A. (2013). Classes of phosphoinositide 3-kinases at a glance. Classes of phosphoinositide 3-kinases at a glance.
Kong, D. and Yamori, T. (2008). Phosphatidylinositol 3‐kinase inhibitors: promising drug candidates for cancer therapy. Phosphatidylinositol 3‐kinase inhibitors: promising drug candidates for cancer therapy.
Kumar, A. and Purohit, R. (2013). Cancer Associated E17K Mutation Causes Rapid Conformational Drift in AKT1 Pleckstrin Homology (PH) Domain. PLoS ONE 8.
Li, T., Christensen, S.D., Frankel, P.H. et al. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California Cancer Consortium trial. Invest New Drugs 30, 741–748 (2012). https://doi.org/10.1007/s10637-010-9562-8
Lindsley, C., Barnett, S., Layton, M. and Bilodeau, M. (2008). The PI3K/Akt Pathway: Recent Progress in the Development of ATP-Competitive and Allosteric Akt Kinase Inhibitors. Current Cancer Drug Targets 8, 7–18.
Loftus, E. V. (2006). Epidemiology and Risk Factors for Colorectal Dysplasia and Cancer in Ulcerative Colitis. Gastroenterology Clinics of North America 35, 517–531.
Mahadevan, D., Chiorean, E. G. and Harris, W. B. et al. (2013). Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies.
Manning, B. D., & Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell, 129(7), 1261–1274. https://doi.org/10.1016/j.cell.2007.06.009
Maurer, M. and Su, T. et al. (2009). 3-Phosphoinositide–Dependent Kinase 1 Potentiates Upstream Lesions on the Phosphatidylinositol 3-Kinase Pathway in Breast Carcinoma.
Meuillet, E. J., Ihle, N., Baker, A. F., Gard, J. M., Stamper, C., Williams, R., Coon, A., Mahadevan, D., George, B. L., Kirkpatrick, L., et al. (2004). In Vivo Molecular Pharmacology and Antitumor Activity of the Targeted Akt Inhibitor PX-316. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics 14, 513–527.
Mundi, P. S., Sachdev, J., McCourt, C., & Kalinsky, K. (2016). AKT in cancer: new molecular insights and advances in drug development. British journal of clinical pharmacology, 82(4), 943–956. https://doi.org/10.1111/bcp.13021
Netland, I.A., Førde, H.E., Sleire, L. et al. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer 16, 657 (2016). https://doi.org/10.1186/s12885-016-2712-4
Pal, S. K., Reckamp, K., Yu, H., & Figlin, R. A. (2010). Akt inhibitors in clinical development for the treatment of cancer. Expert opinion on investigational drugs, 19(11), 1355–1366. https://doi.org/10.1517/13543784.2010.520701
Raatschen, H. J., Simon, G. H., Fu, Y., Sennino, B., Shames, D. M., Wendland, M. F., McDonald, D. M., & Brasch, R. C. (2008). Vascular permeability during antiangiogenesis treatment: MR imaging assay results as biomarker for subsequent tumor growth in rats. Radiology, 247(2), 391–399. https://doi.org/10.1148/radiol.2472070363
Rayburn, E. R., Ezell, S. J., & Zhang, R. (2009). Anti-Inflammatory Agents for Cancer Therapy. Molecular and cellular pharmacology, 1(1), 29–43. https://doi.org/10.4255/mcpharmacol.09.05
Richardson, P. G., Eng, C., Kolesar, J., Hideshima, T., & Anderson, K. C. (2012). Perifosine , an oral, anti-cancer agent and inhibitor of the Akt pathway: mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert opinion on drug metabolism & toxicology, 8(5), 623–633. https://doi.org/10.1517/17425255.2012.681376
Rothman, I., Stanford, J. L., Kuniyuki, A. and Berger, R. (2004). Self-report of prostatitis and its risk factors in a random sample of middle-aged men.
Sale, E. M. and Sale, G. J. (2007). Protein kinase B: signalling roles and therapeutic targeting. Cellular and Molecular Life Sciences 65, 113–127.
Sarkar, D. and Fisher, P. B. (2006). Molecular mechanisms of aging-associated inflammation. Cancer Letters 236, 13–23.
Sato, S., Fujita, N. & Tsuruo, T. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 21, 1727–1738 (2002). https://doi.org/10.1038/sj.onc.1205225
Sayle KL, Bentley J, Boyle FT, Calvert AH, Cheng Y, Curtin NJ et al. (2003). Structure-based design of 2-arylamino-4-cyclohexylmethyl- 5-nitroso-6-aminopyrimidine inhibitors of cyclin-dependent kinases 1 and 2. Bioorg Med Chem Lett 13: 3079–3082.
Singh, A.R., Joshi, S., George, E. et al. Anti-tumor effect of a novel PI3-kinase inhibitor, SF1126, in 12 V-Ha-Ras transgenic mouse glioma model. Cancer Cell Int 14, 105 (2014). https://doi.org/10.1186/s12935-014-0105-9
Strukov AI, Paukov VS, Orekhov OO. [Morphology, pathogenesis and classification of interstitial lung diseases] Arkhiv Patologii. 1984 ;46(7):3-14.
Sun, H., Zhu, A., Zhou, X., & Wang, F. (2017). Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget, 8(32), 52642–52650. https://doi.org/10.18632/oncotarget.16991
Tolcher AW, Yap TA, Fearen I, Taylor A, Carpenter C, Brunetto AT, et al. A phase I study of MK‐2206, an oral potent allosteric AKT inhibitor (AKTi), in patients (pts) with advanced solid tumor (ST). J Clin Oncol 2009, American Society of Clinical Oncology Meeting Abstract
Vane, J. R. and Botting, R. M. (1998). Mechanism of action of anti-inflammatory drugs: an overview. Selective COX-2 Inhibitors 1–17.
Vara, J. Á. F., Casado, E., Castro, J. D., Cejas, P., Belda-Iniesta, C. and González-Barón, M. (2004). PI3K/Akt signalling pathway and cancer. Cancer Treatment Reviews 30, 193–204.
Whale, A., Colman, L., Lensun, L. et al. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Sig Transduct Target Ther 2, 17063 (2017). https://doi.org/10.1038/sigtrans.2017.63
Xie, J., Wang, X., & Proud, C. G. (2016). mTOR inhibitors in cancer therapy. F1000Research, 5, F1000 Faculty Rev-2078. https://doi.org/10.12688/f1000research.9207.1
Zentella-Dehesa, A. and García-Román, J. (2013). Vascular permeability changes involved in tumor metastasis.
Zhang, H., Dou, J., Yu, Y., Zhao, Y., Fan, Y., Cheng, J., Xu, X., Liu, W., Guan, S., Chen, Z., shi, Y., Patel, R., Vasudevan, S. A., Zage, P. E., Zhang, H., Nuchtern, J. G., Kim, E. S., Fu, S., & Yang, J. (2015). mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis : an international journal on programmed cell death, 20(1), 50–62. https://doi.org/10.1007/s10495-014-1066-0
Zhang, L., Li, Y., Wang, Q. et al. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol Cancer 19, 10 (2020). https://doi.org/10.1186/s12943-019-1112-1
Zhu J, Huang J-W, Tseng P-H, Yang Y-T, Fowble J, Shiau C-W et al. (2004). From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Research 64: 4309–4318.